


- Jan 24, 2022
- 3 min read
Updated: Jan 24, 2022
The paper was published in Nanoscale (2022).
Graphene oxide (GO) attracts significant attention due to its easily scalable and low-cost synthesis. GO was suggested as an attractive material for numerous applications in sensing, energy storage, 2D electronics and optoelectronics, photocatalysis, and memristors. This great tailorability of GO properties is apparently due to a rich variety of possible chemical compositions and structures. To further extend GO practical applications, it is highly important for the whole GO community to sort out the connections between a chemical composition, an atomic structure and electronic properties of GO.
In this paper we presented the results of our complex computational study of graphene oxide as a material with widely tunable structure and electronic properties. Using density-functional calculations, we considered various phases of fully oxidized GO and determined the most favorable structures among them. For the three selected stoichiometries, armchair and zigzag graphene nanoroads were paved in GO, and their thermodynamical, electronic, and magnetic properties were thoroughly studied for a broad range of nanoroad widths.
We showed that graphene nanoroads form highly stable, low-energy interfaces with GO and demonstrate the non-trivial dependence of electronic properties on a nanoroad width. Armchair nanoroads (Figure (a)) are characterized by the oscillating decrease of the band gap when widening a nanoroad (Figure (b)), and the same trend was observed for the charge carriers’ effective masses with their lowest values being in the range 0.05 – 0.2me for wide enough nanoroads (Figure (b), inset) that sounds promising for applications of patterned GO as a 2D semiconductor. Our results provide a possible explanation for the wide range of GO band gaps observed experimentally.
The electronic properties of zigzag nanoroads (Figure (c)) were found to be intimately connected to their magnetic state. Non-magnetic and ferromagnetic states are metastable and lead to a metallic conductivity, whereas antiferromagnetic arrangement was found to be a semiconducting ground state for zigzag nanoroads (Figure (d)). We have also demonstrated that AFM zigzag nanoroads can change their conductivity from a semiconducting to a half-metallic type when subjected to the external electric field.
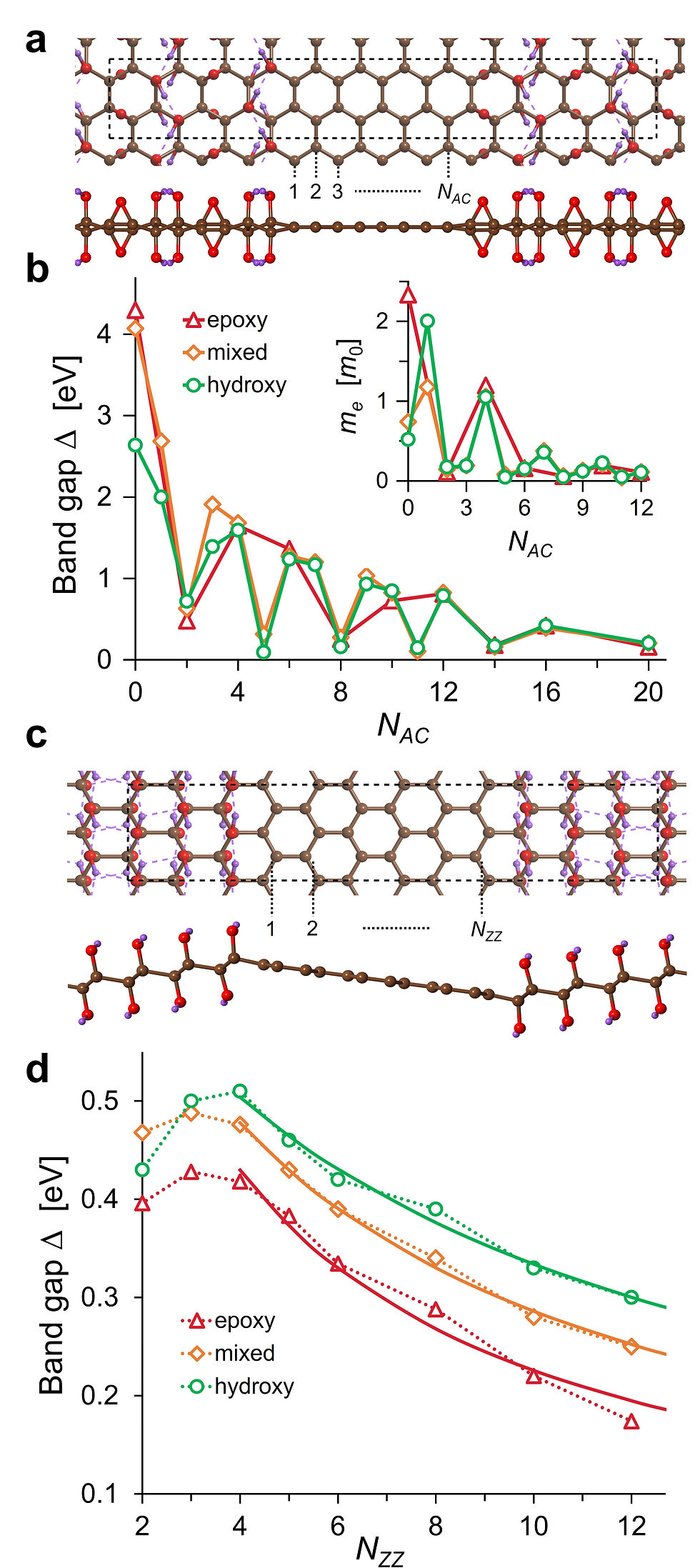
a) The atomic structure of 8-mixed-AGONR’ (top and side views are given). The dashed box shows the orthorhombic unit cell used in calculations. The numbers (1, 2, 3, …, NAC) indicate the number of dimer lines constituting the nanoroad width. b) The dependence of AGONR’ band gap Δ on the nanoroad width NAC for the three considered GO compositions: epoxy-GO (red), mixed GO (orange), and hydroxy-GO (green). The inset shows effective masses and electrons as the function of AGONR’ index. c) The atomic structures of 6-hydroxy-ZGONR’ (top and side views). The dashed boxes denote the orthorhombic unit cells used in calculations. The numbers (1, 2, …, NZZ) mark zigzag carbon chains making up a graphene nanoroad. d) The dependence of antiferromagnetic ZGONR’ band gap Δon the nanoroad width NZZ. The fitting (starting from NZZ = 4) with the hyperbolic law is shown with solid lines of corresponding colors
All mentioned effects are manifested very similarly for all three GO stoichiometries, which allows us to conclude that the electronic properties of GONR’ are mainly determined by graphene regions and weakly depend on the exact chemical composition of the GO matrix. In principle, this should relieve experimentalists from the need to thoroughly control the composition of the synthesized patterned GO without losing its remarkable electronic properties. Together with its cheapness and wide availability, this means that GO could finally become a material where graphene nanoroads with their exciting electronic properties can be observed and measured experimentally.
We believe that using state-of-the-art experimental techniques, such as STM, electron beam irradiation and nanolithography, it should be possible to create graphene nanoroads. Although currently there are no direct evidences for graphene oxide, we believe that these methods successfully applied to hydrogenated and fluorinated graphene can be generalized onto the case of oxygen groups as well. We assume that our theoretical predictions may serve as a good motivation for experimentalists to go in this direction, since the possibility to transform GO into a semiconducting structure demonstrated in our work has a great potential for practical applications. We hope that our results would be useful for people working in the GO community and beyond.

